Molecular Biology Explained

A living organism must maintain itself continuously by producing the substances it needs to function. Proteins are one of these key substances and come in a seemingly infinite variety of shapes and structures. They are responsible for virtually everything that goes on in your body and cells, and are produced every second of an organism’s life. They are needed for various functions such as cell growth, tissue repair, signalling between cells, immune responses, muscle movement, enzymes to help carry out chemical reactions, hormones, and they also act as the building blocks for structures such as hair, feathers, insect shells and collagen.
The template for making proteins
Despite such huge variety, proteins are mostly defined by their sequence of amino acids linked together into ‘polypeptide chains’.
The template that codes for these proteins is a molecule called DNA – deoxyribonucleic acid.The central dogma- or more precisely hypothesis- of molecular biology states that genetic information translates from nucleic acid to protein, and not the other way round. This means that DNA makes RNA, and RNA makes proteins.There are exceptions to this process, such as when certain viruses are able to make DNA copies of their own RNA genome. But in most organisms, DNA is copied to RNA. The process by which DNA is copied to RNA is called transcription, and that by which RNA is used to produce proteins is called translation. This is the reason why DNA is such a key part of every organism’s vital processes.
Deoxyribonucleic Acid (DNA)
 In humans, the nucleus of each cell contains 3 billion base pairs of DNA distributed over 23 pairs of chromosomes, and each cell has two copies of the genetic material. This is known collectively as the human genome. The human genome contains around 30 000 genes, each of which codes for one protein.
In humans, the nucleus of each cell contains 3 billion base pairs of DNA distributed over 23 pairs of chromosomes, and each cell has two copies of the genetic material. This is known collectively as the human genome. The human genome contains around 30 000 genes, each of which codes for one protein.
The deoxyribose nucleic acid molecule consists of a sugar molecule connected to one of four bases – guanine, cytosine, adenine and thymine. A Base joined to a sugar is called a nucleotide. Phosphate molecules join the ribose sugars together to form a chain , one side of the DNA double helix.
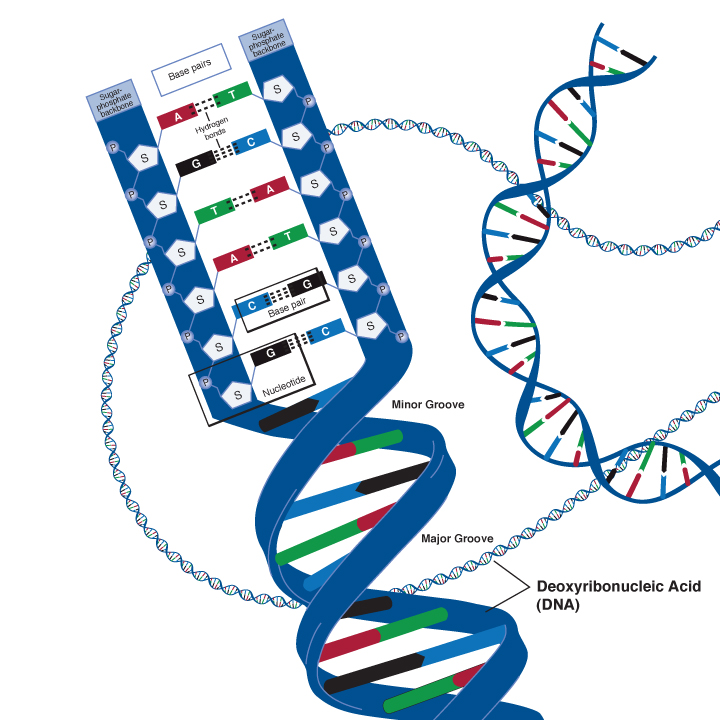
The four bases have different unique shapes which mean that they will only ever fit together in specific ways, like pieces of a puzzle. Adenine will only ever pair with Thymine, and Cytosine will only ever pair with Guanine. This means that the two strands of DNA are complementary, and effectively, are templates of each other.
Transcription
To see how DNA is used to make proteins, we need to introduce its sister compound Ribonucleic acid (RNA). RNA differs to DNA in a number of ways. The most important of these differences are firstly, that it is a single strand, rather than a double strand. And secondly, instead of Thymine, it has Uracil as a base.
Transcription is the process by which DNA is copied (transcribed) to messenger RNA (mRNA), which carries the information needed for protein synthesis.
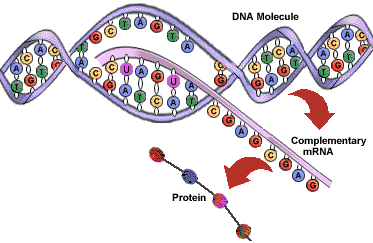 So when a certain protein needs to be manufactured, the part of the DNA double helix which contains the relevant gene for that protein unzips, exposing the sequence of bases of A,C, G or Ts.
So when a certain protein needs to be manufactured, the part of the DNA double helix which contains the relevant gene for that protein unzips, exposing the sequence of bases of A,C, G or Ts.
Then with the help of enzymes, free-floating nucleotides of mRNA link up with their corresponding nucleotides of DNA, and become joined together in a single strand of mRNA nucleotides.
Once the whole gene has become transcribed into a strand of mRNA, the mRNA moves off, and the DNA is zipped back up.
Translation
Now that the genetic code has been copied into mRNA, the code then needs to be translated to become a protein, and as mentioned earlier, all a protein really is, is a sequence of amino acids.
To do this, the mRNA is fed into a tiny molecular ‘machine’, called a Ribosome, which helps match up the correct amino acid for each part of the mRNA code. This is done for every three bases in the RNA sequence, called a codon. This three base sequence, gives a variety of different code combinations, each of which corresponds to a particular amino acid. So as the ribosome runs along the mRNA sequence, the correct amino acids are brought together in the correct sequence, and linked together to become a specific protein.
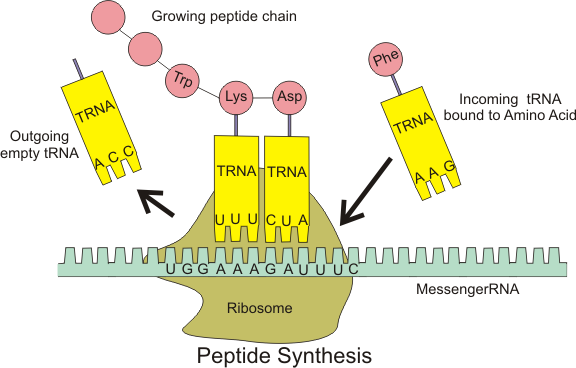
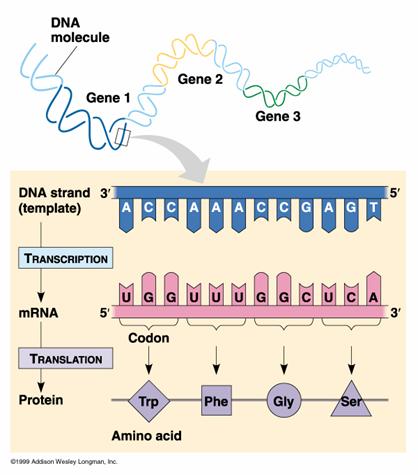
Our lab experiments



A lot of the experiments we run at the London BioHackspace are gene-typing experiments, which is essentially looking at our DNA and testing for the presence of certain genes.
For example, one of our projects is a test for certain blood types. We can do this because your blood group is determined by a certain combination of antigens in your red blood cells. Because antigens are proteins, coded in your DNA, we should be able to tell which antigens and which blood type you are, just by looking at your DNA.
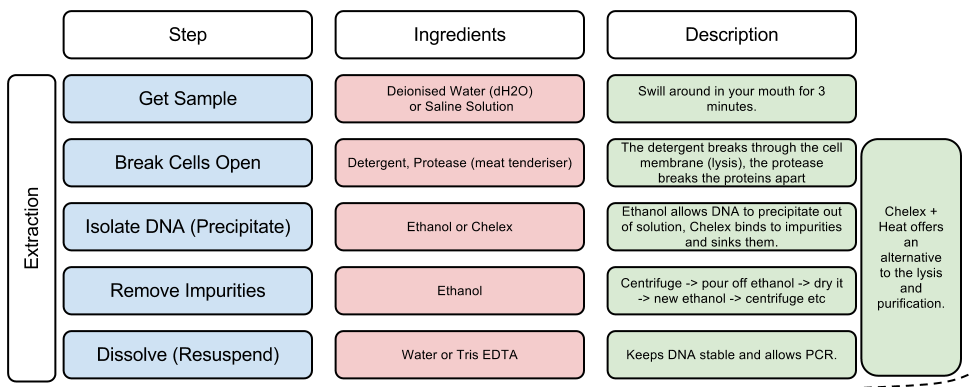
DNA extraction
But first we need to get the DNA out. Not only is DNA locked in our cells, deep inside its nucleus, DNA is also tightly wrapped up with proteins. So we first need to take a collection of our own cells, normally by lightly rubbing the inside of our cheeks, and then we break open the collected cells, and extract the DNA using a number of chemical processes, which are outlined in the table below:
Polymerase Chain Reaction (PCR)
After breaking down the cells, and extracting the DNA, we now need to identify the gene we’re looking for. But there are a couple of problems. Firstly, we have now collected our entire genome of DNA, and not just the gene we are looking for. And secondly, it is such a small sample of DNA that we’re unlikely to be able to see anything at all. So we need to make lots of copies of the specific section of DNA we’re looking for, and we do this using a method called Polymerase Chain Reaction (PCR).
This is the exact same process that gets used with forensics, and is how they are able to match the DNA of a suspect, from just a small sample of hair.
To do PCR, we need to add a few ingredients to the tube, and then repeat a specific cycle of heating and cooling to get the DNA to replicate. This is done in a machine called a thermal cycler.
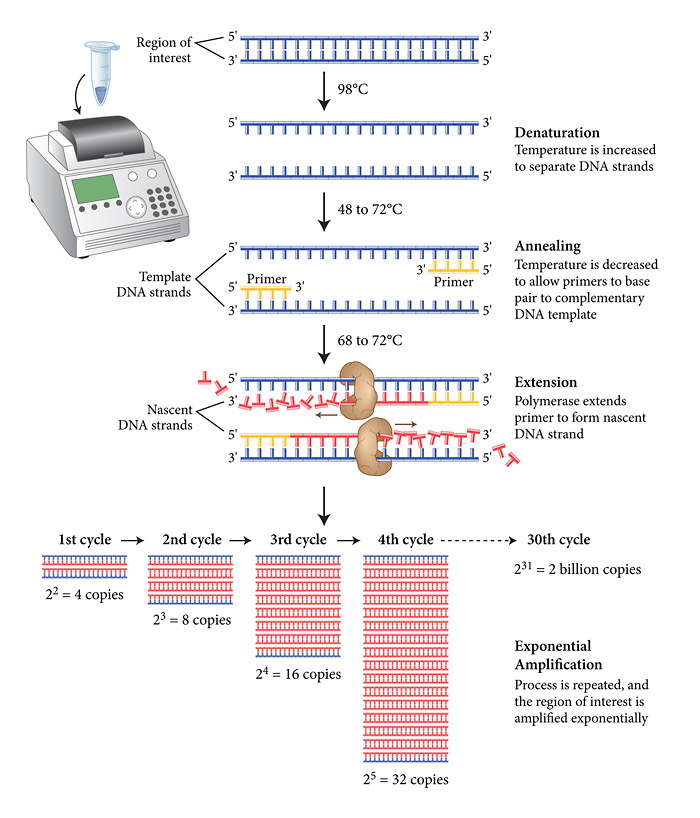
The thermal cycler will go through many cycles of heating the solution through different temperatures. The first stage involves heating to the hottest temperature, 96°C, which will cause the double DNA strands to separate. Then it will be cooled to allow the process of copying the DNA to begin.
In order to choose the gene we want to copy, we use primers. Primers are short pieces of DNA that are made in a laboratory. Since they’re custom built, primers can have any sequence of nucleotides you’d like.
In a PCR experiment, we use two primers which are designed to match the beginning and the end of the segment of DNA we want to copy. And in the case of our blood typing experiment, we’re using primers that match the beginning of the gene that codes for the blood antigens.
In the annealing stage, through complementary base pairing, one primer attaches to the top strand at one end of your segment of interest, and the other primer attaches to the bottom strand at the other end.
Then we heat the solution up to allow extension to begin. To copy the remaining sequence of DNA, we use an enzyme called DNA Polymerase. DNA Polymerase is a naturally-occurring enzyme whose function is to copy a cell’s DNA before it divides in two. When a DNA polymerase molecule bumps into a primer that’s base-paired with a longer piece of DNA, it attaches itself near the end of the primer and starts adding nucleotides.
Human DNA polymerase would break down at the temperatures we’re using in our PCR experiment. So the DNA polymerase that’s most often used in PCR comes from a strain of bacteria called Thermus aquaticus that live in the hot springs of Yellowstone National Park. It can survive near boiling temperatures and works quite well at 72°C.
In our PCR tube we’ve already added a mixture of four types of nucleotides found in DNA – A’s, C’s, G’s and T’s. So in the extension process, the DNA polymerase grabs nucleotides that are floating in the liquid around it and attaches them to the end of a primer, until the whole gene has been copied, and the end of a cycle is reached.
Then the process repeats all over again, with the two newly formed double strands of DNA themselves splitting, and being replicated. And so on… Until we have a highly concentrated solution of DNA for our blood type gene.
Gel electrophoresis
In the tube, even though we’ve successfully copied lots of fragments of DNA which are of the blood type gene, there are many other fragments left over from the PCR reaction. So now we want to be able to separate all of the various fragments of DNA in the tube, so that we can identify the blood typing gene fragment, and we do this by size.
To do this we use a process called Gel Electrophoresis. We make a gel out of a chemical called agarose, through which solutions of DNA are able to move, and we place our solutions of DNA from the PCR process into some wells which are indented in the gel. The gel also contains a chemical called Ethidium Bromide, which binds with the DNA, and acts as a fluorescent tag, which will allow us to see the DNA at the next stage.
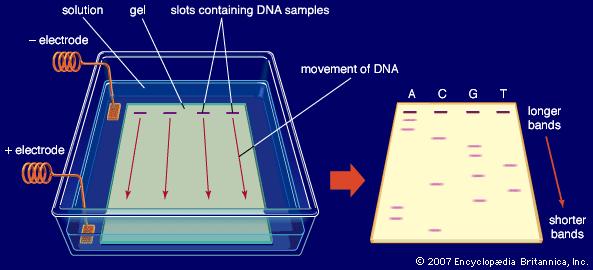
The gel is placed in a solution and an electric current is passed through. Because DNA is a negatively charged molecule, as this current is applied, the strands of DNA will move slowly towards the positive electrode. However, the strands of DNA will move at different speeds. Shorter strands of DNA (for example short leftover chunks from the PCR reaction) find it easier to move through the gel, and so will move along much quicker. Whereas longer strands of DNA (e.g. gene sequences) will move more slowly through the gel. So after running the gel for a certain period of time, the different sized strands of DNA will be separated, appearing at different points along the gel.
 But for us to find out exactly how long these DNA strands are, we need reference markers, which we call a ladder. This is a solution containing a mixture of DNA fragments of certain sizes (e.g. 300 nucleotides long, 500, 700, 1,000 etc.). When the ladder solution is placed into a well next to one containing our PCR DNA sample, it will run down the gel in the same way, with the various different sized chunks of DNA appearing at different positions down the gel. So if our PCR DNA sample, appears next to the ladder band representing 500 bp, we know our sample contains DNA of a gene roughly 500 nucleotides long. Or if it appears halfway between the bands of 1,500 and 2,000, we can make an estimate of it being roughly 1,750 nucleotides long.
But for us to find out exactly how long these DNA strands are, we need reference markers, which we call a ladder. This is a solution containing a mixture of DNA fragments of certain sizes (e.g. 300 nucleotides long, 500, 700, 1,000 etc.). When the ladder solution is placed into a well next to one containing our PCR DNA sample, it will run down the gel in the same way, with the various different sized chunks of DNA appearing at different positions down the gel. So if our PCR DNA sample, appears next to the ladder band representing 500 bp, we know our sample contains DNA of a gene roughly 500 nucleotides long. Or if it appears halfway between the bands of 1,500 and 2,000, we can make an estimate of it being roughly 1,750 nucleotides long.
Gel visualisation
Once the gel has run for long enough, we switch off the current, and then place the gel under UV light. The fluorescent marker provided by the Ethidium Bromide we put in at the start, means that the DNA will glow quite clearly under the UV.
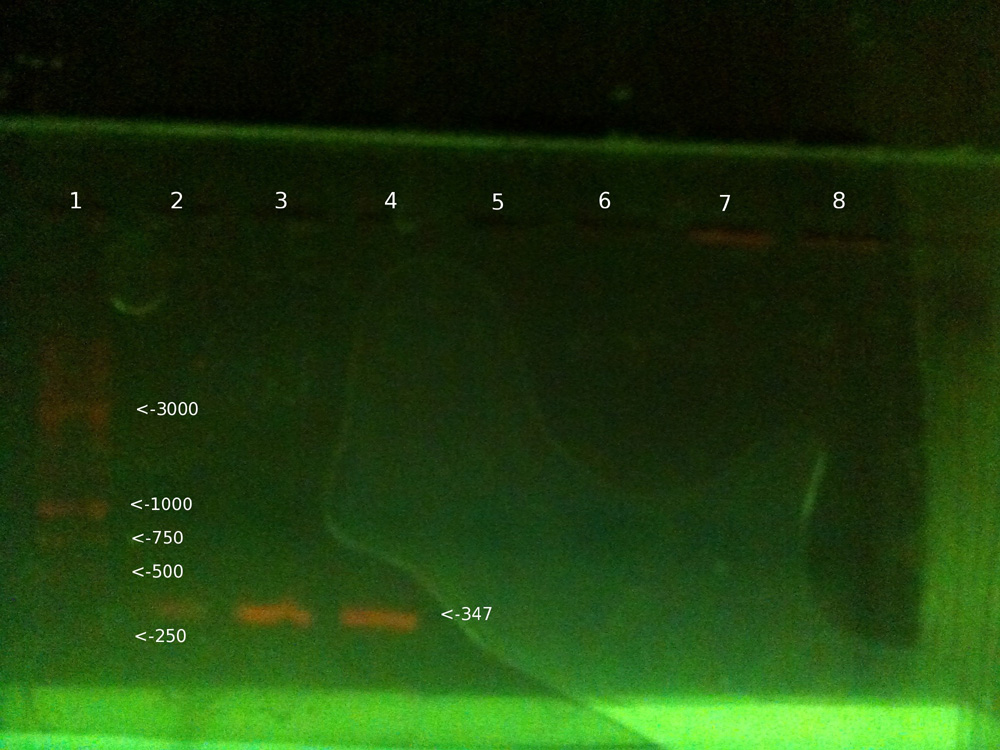
As you can see, some clear bands of DNA appear roughly around the 347 base pair mark, which is the length of the DNA sequence we are looking for involved in determining blood group. Once we have this section, we add Alu1 restriction enzyme, which cuts the sequence if it is a blood group B allele, but not if it’s A or O.


